Mukopolisakkaridoz glikozaminoglikanların depolanmasına dayanan lizozomal depo hastalıkları için toplu bir terimdir. Tüm hastalıklar benzer semptom ve formlar geliştirir. Sendromların şiddeti büyük ölçüde değişir.
Mukopolisakkaridoz nedir?

© Sebastian Kaulitzki - stock.adobe.com
bir Mukopolisakkaridoz tek bir hastalık olarak yok. Mukopolisakkaridoz terimi, çok sayıda depo hastalığı için toplu bir terimdir ve bunlar, hücrelerin lizozomlarında glikozaminoglikanların (GAG) depolanma bozukluklarına dayanmaktadır. Depolama, bağlantıların sonlandırılması çalışmadığı için aşamalı olarak gerçekleşir.
Tüm mukopolisakkaridozlar genetiktir. Her hastalık, karşılık gelen GAG'nin parçalanmasını katalize eden belirli bir enzime sahip değildir. Tüm mukopolisakkaridozlar çok nadir hastalıklardır ve sıklıkla benzer seyirler gösterirler. Tedavi edilmezse, sürekli artan tortular hücreleri yok eder. İşlem sırasında organlar yok edilir. Hastalık hem bebeklik döneminde hem de yetişkinlikte başlayabilir.
Mukopolisakkaridoz, dört farklı glikozaminoglikan grubundan kaynaklanabilir:
- Heparan sülfat
- Keratan sülfat
- Kondroitin sülfat
- Dermatan sülfat.
Tüm glikozaminoglikanlar, bir proteine bağlı bir polisakkarit zincirinden oluşur. Karbonhidrat bileşeni moleküler kütlenin yüzde 95'ini, protein bileşeni ise yüzde beşini oluşturur. Hangi glikozaminoglikanın ve hangi enzimin etkilendiğine bağlı olarak, mukopolisakkaridozlar altı farklı ana forma ayrılabilir: Bunlar Hurler / Scheie hastalığı (MPS I), Hunter's hastalığı (MPS II), Sanfilippo hastalığı (MPS III), Morquio hastalığı ( MPS IV), Maroteaux-Lamy hastalığı (MPS VI) ve Sly hastalığı (MPS VII). Tüm türlerin şiddetli ve hafif formları vardır.
nedenleri
Tüm mukopolisakkaridozların nedeni, hücrelerin lizozomlarında glikozaminoglikanların (GAG) depolanmasının artmasıdır. Karşılık gelen biyopolimerlerin bozunması bozulur. Her bir bozukluk için ya belirli bir enzim eksiktir ya da bu enzim yanlış çalışıyor. Her enzim için birkaç mutasyon olabilir. Karşılık gelen mutasyonun kalıtımı otozomal resesif, otozomal dominant veya x'e bağlı resesif olabilir.
Enzimatik bir işlem genellikle birkaç reaksiyon aşamasını içerdiğinden, aynı glikozaminoglikan için birkaç enzim teorik olarak mutasyona uğratılabilir. Bozukluğun semptomları aynı veya benzer olacaktır.
- at MPS IHurler veya Scheie hastalığı, alfa-l-iduronidaz enzimi kusurludur.
- MPS II kusurlu bir iduronat-2-sülfataz ile Hunters Sendromunu temsil eder.
- Sanfilippo Sendromu (MPS III) birkaç alt türe ayrılabilir. Bu durumda birkaç enzim etkilenebilir.
- Morquio hastalığı (MPS IV) kusurlu bir-galaktosidazdan kaynaklanır.
- Maroteaux-Lamy sendromunda (MPS VI) N-asetil-galaktozamin-4-sülfat sülfatazdır.
- Sly hastalığı (MPS VII) kusurlu β-glukuronidazdan kaynaklanır. Karşılık gelen glikozaminoglikanlar lizozomlarda depolandığında, bunlar daha da büyür.
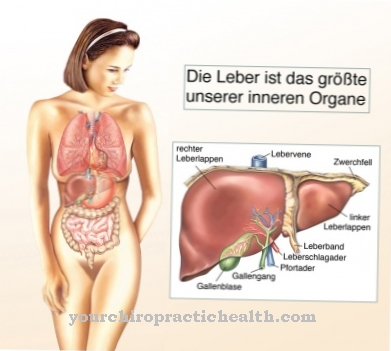
Hücreler de bozunmamış GAG'ler için giderek daha fazla alana ihtiyaç duydukları için büyür. Bu aynı zamanda birçok organın büyümesinde de belirgindir. Tipik bir semptom, karaciğer ve dalağın sürekli büyümesidir. Tedavi edilmezse, depo hastalıkları organların kademeli olarak tahrip edilmesiyle ölüme yol açar.
Belirtiler, rahatsızlıklar ve belirtiler
Semptomlar tüm hastalıklar için benzerdir. Şiddetli ve hafif formlar var. Bununla birlikte, hafif bir seyir sadece hastalığın daha yavaş ilerlediği anlamına gelir. Son kurs her zaman aynıdır. İskelet sisteminde ilerleyici deformasyon, eklem kontraktürleri, yüz özelliklerinde kabalaşma ve karaciğer ve dalakta genişleme vardır.
Kısa veya uzun vadede zihinsel ve motor beceriler azalır. Hastalıkların şiddetli formlarında klinik tablolar çok benzerdir. Göbek ve kasık fıtıkları, kalp sorunları ve solunum yolu enfeksiyonları erken dönemde ortaya çıkar. Zamanla solunum yollarının daralması ve bademciklerin ve bademciklerin büyümesi yoğun uyku apnesi problemlerine dönüşür.
Tanı ve hastalık seyri
Mukopolisakkaridozlar, idrarın atılan glikozaminoglikanlar açısından incelenmesiyle teşhis edilebilir. Mukopolisakkaridozda değerler her zaman artar. Lökositler veya fibroblastlardaki şüpheli kusurlu enzimin aktivitesi de belirlenebilir. Glikozaminoglikanların belirli bir atılım modeli şüpheyi karşılık gelen bir enzime götürür ve ardından incelenir.
Komplikasyonlar
Mukopolisakkaridozun bir sonucu olarak, etkilenenler çeşitli şekil bozuklukları ve iskelet şikayetlerinden muzdariptir. Hastanın günlük yaşamını önemli ölçüde sınırlayabilecek deformasyonlar meydana gelir. Kural olarak, eklemler de mukopolisakkaridozdan etkilenir, böylece hastanın hareketi kısıtlanır.
Özellikle çocuklar etkilenir ve ciddi şekilde gecikmiş gelişimden muzdariptir, böylece yetişkinlikte de çeşitli dolaylı zararlar meydana gelebilir. Mukopolisakkaridozun kalpte veya solunumda sorunlara neden olması nadir değildir. En kötü senaryoda, ani kardiyak ölüm, ilgili kişinin ölümüne yol açabilir. Solunum güçlüğü nedeniyle hastalar yorgunluk ve halsizlik çekerler.
Etkilenenlerin dayanıklılığı da büyük ölçüde azalır. Geceleri nefes alma zorluklarının uyku problemlerine ve dolayısıyla depresyona yol açması nadir değildir. Mukopolisakkaridoz, hastanın yaşam kalitesini önemli ölçüde düşürür. Bu hastalığın nedensel tedavisi maalesef mümkün değildir. Bu nedenle etkilenenler semptomları tedavi etmek için kemik iliği donörlerine bağımlıdır. Belirli bir komplikasyon yok. Bununla birlikte, çoğu durumda, hastalar yaşam boyu tedaviye bağımlıdır.
Ne zaman doktora gitmelisiniz?
Vücut yapısındaki değişiklikler ve anormallikler bir sağlık bozukluğuna işaret eder. Kalıcı optik özellikler ortaya çıktığında veya ilgili kişi kasıtlı olarak duruşunu optimize etmekte sorun yaşadığında bir doktor ziyareti gereklidir.Eklemlerde şişlik, yüz hatlarında değişiklikler veya göğüste büyüme, bir doktor tarafından yoğun bir şekilde muayene edilmeli ki teşhis konulmalıdır. Hareket olasılıklarında kısıtlamalar, günlük gönüllü kontrolde düzensizlikler ve fiziksel ve zihinsel performansta azalma varsa bir doktor gereklidir. Gece uykusu sırasında kalp ritminde bozukluk, nefes almada sorun veya kesinti olması durumunda bir doktora başvurun.
Boğazda şişme, boğazda gerginlik hissi, yutkunma hareketinde rahatsızlıklar ve seslendirme değişiklikleri endişe verici kabul edilir. Semptomların hafifletilebilmesi için bir doktor tarafından muayene edilmeleri gerekir. İlgili kişi daha fazla enfeksiyona maruz kalırsa, konsantre olma ve dikkat etme yeteneği azalırsa veya tekrar tekrar göbek veya kasık fıtıkları oluşursa, gözlemler bir doktora bildirilmelidir.
Ani lekeler, ciltte sararma ve iç huzursuzluk muayene edilmeli ve tedavi edilmelidir. Vücutta ağrı, düşük yaşam kalitesi ve davranış sorunları olur olmaz doktora ihtiyaç duyulur. Nefes darlığı riski varsa ambulans servisi gereklidir. Bu akut durumu önlemek için olabildiğince erken bir doktora danışılmalıdır.
Terapi ve Tedavi
Nedensel bir terapi bugün henüz mümkün değil. Bununla birlikte, araştırma projelerinde bu hastalıklar için gelecekteki gen tedavisine yönelik bazı yaklaşımlar vardır. Ne yazık ki, şu anda bu alanda somut bir sonuç yok. Bununla birlikte, Hurler hastalığı için gen terapisi üzerine bir klinik araştırma Barselona'da başlayacak. Bazı mukopolisakkaridoz formlarında, kemik iliği transferlerinin bireysel vakalarda etkili olduğu kanıtlanmıştır. Bu, örneğin, Hunter hastalığı, Hurler hastalığı veya Sanfilippo hastalığı ile ilgilidir.
Bu kemik iliği transferi yoluyla, hastalıklı kök hücreler bir donörden alınan sağlıklı kök hücreler ile değiştirilir. Bu, organizmanın eksik enzimi yeterince geri kazanmasını sağlar. Enzim replasman tedavisi de birçok durumda karşılığını verir. Ancak bu replasman tedavisi ömür boyu uygulanmalıdır. Bununla birlikte, umut verici tedavilerin artık mümkün olmadığı durumlar da vardır. Ancak burada önemli olan semptomatik tedavilerin yapılmasıdır.
İlaçlarınızı burada bulabilirsiniz
➔ Ağrı kesici ilaçlarGörünüm ve tahmin
Mukopolisakkaridozlu hastalarda daha fazla gelişme ayrı ayrı değerlendirilmelidir. Bu terim, çeşitli depo hastalıkları için toplu bir terimdir. Bunlar her hastada farklı derecelerde mevcuttur ve yoğunlukları ayrı ayrı ifade edilir. Tıbbi bakım uygulanmazsa, etkilenen tüm kişilerin iç organları yaşamları boyunca kademeli olarak yok edilir. Bu, ortalama beklenen ömrün kısalmasıyla sonuçlanır.
Erken teşhisle, kişisel olarak optimize edilmiş bir terapi uygulanabilir. Bu, sağlık gereksinimleri ve hastanın mevcut şikayetleri ile bağlantılıdır. Sağlıkta istikrarlı bir iyileşme sağlamak için temelde uzun vadeli tedavi gereklidir. Her biri farklı riskler ve yan etkilerle ilişkili cerrahi müdahaleler meydana gelebilir. Operasyon başka bir komplikasyon olmaksızın devam ederse, genellikle sonrasında semptomlar düzelir.
Yine de hayatın akışı içinde istenmeyen gelişmeler ve aksaklıklar yaşanabilir. Bazı durumlarda, yalnızca bir kemik iliği nakli genel yaşam kalitesini iyileştirebilir. Genel koşullar nedeniyle, hasta güçlü bir duygusal ve zihinsel yük yaşar. Semptomlar nedeniyle normal günlük yaşam genellikle mümkün değildir. Psikolojik komplikasyonlar ortaya çıkabilir ve durumun daha da kötüleşmesine yol açabilir.
önleme
Mukopolisakkaridozlar kalıtsal hastalıklar olduğu için önlenmesi mümkün değildir. Mevcut bir hastalık durumunda, zamanında tedavi ile tedavinin başarısı sağlanabilir. Ek olarak, akciğer ve kalp fonksiyonunun sürekli izlenmesi gereklidir. Ailede daha önce mukopolisakkaridoz vakaları meydana gelmişse, aile çocuk sahibi olmak istiyorsa, genetik danışmanlık yoluyla hastalık riski değerlendirilebilir.
tamamlayıcı tedavi
Mukopolisakkaridoz durumunda, çoğu durumda hasta, takip bakımı için yalnızca birkaç seçeneğe sahiptir, bu nedenle etkilenen kişinin öncelikle erken bir aşamada doktora başvurması gerekir. Ancak bu hastalığın erken teşhisi ve tedavisi ile daha fazla komplikasyon önlenebilir, böylece ilk belirti ve semptomlar ortaya çıkar çıkmaz bir doktora başvurulmalıdır.
Çoğu durumda, etkilenenler, semptomları hafifletip sınırlayabilen cerrahi müdahalelere bağımlıdır. Ancak mukopolisakkaridoz genetik bir hastalık olduğu için genellikle tam olarak tedavi edilemez.
Bu nedenle ilgili kişi çocuk sahibi olmak istiyorsa bu hastalığın çocuklarda tekrarını önlemek için öncelikle bir doktora başvurmalıdır. Tedavi sırasında aile desteğinin olması da genellikle çok önemlidir. Bu aynı zamanda depresyonu ve diğer psikolojik rahatsızlıkları da önleyebilir. Mukopolisakkaridoz, etkilenen kişinin yaşam beklentisinin azalmasına neden olabilir, bu nedenle ilerideki gidişat büyük ölçüde tanı zamanına bağlıdır.
Bunu kendin yapabilirsin
Mukopolisakkaridozda kendi kendine yardım olanakları, semptomları hafifletmek ve böylece yaşam kalitesini artırmakla sınırlıdır. Kendi kendine yardım gruplarının çok yararlı olduğu kanıtlanmıştır, çünkü diğer ebeveynlerle yapılan değişim değerli ipuçları ortaya çıkarır ve çoğu zaman korkuları ve endişeleri hafifletebilir ve geleceğe daha olumlu bir bakış açısı sağlayabilir.
Ev ortamında sıklıkla derinleştirilebilen fizyoterapi, uğraşı terapisi, konuşma terapisi ve diğer terapi türlerine eşlik etmek artık yaşamın ayrılmaz bir parçası.
Kendiniz ve etkilenen çocuk için hayatı olabildiğince kolaylaştırmak için, yaşam ortamını mümkün olduğunca erken engelliler için erişilebilir hale getirmeniz önerilir. Çocuğun yaşı ve kilosu arttıkça, yüksekliği ayarlanabilir bakım yatakları, bakıcı için büyük bir fiziksel rahatlama olduğunu kanıtlıyor. Epilepsi uyarı cihazları ve diğer teknik yardımlar, geceleri bile mümkün olan en iyi güvenliği sağlar ve daha rahat uyuyabilmeleri için ebeveynleri geceleri rahatlatır.
Bir semptom günlüğü tutmak, doktorun yeni semptomları tanımasına ve muhtemelen mevcut semptomların tedavisini düzeltmesine yardımcı olabilir, çünkü ilaç tedavisi genellikle istenen etkiyi değil, tersi etkiyi gösterir.
Hastalık akrabaları için çok zor olduğundan, pillerini şarj etmek için kendilerine çok az yer açmaları gerekir. Bu tedaviler, önleyici bakım veya daha sonra bakımevinde bir tatil içerebilir.
.jpg)

.jpg)
.jpg)


.jpg)





.jpg)
.jpg)